
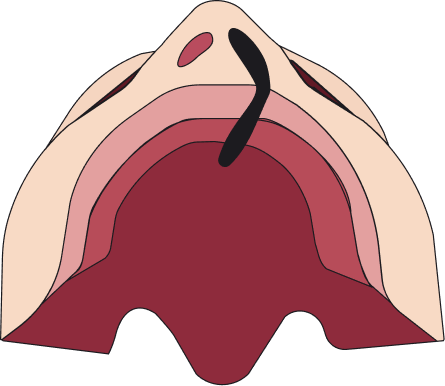
Unilateral incompleto
Afecta solo un lado del labio superior y no se extiende hasta la nariz. Generalmente afecta la parte más baja del labio; no involucra el paladar.

Las fisuras son alteraciones del desarrollo de la cara que se producen en etapas muy precoces de la gestación.
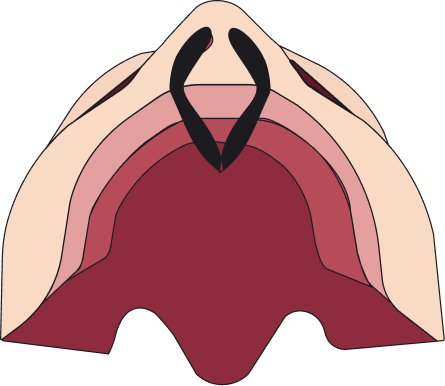
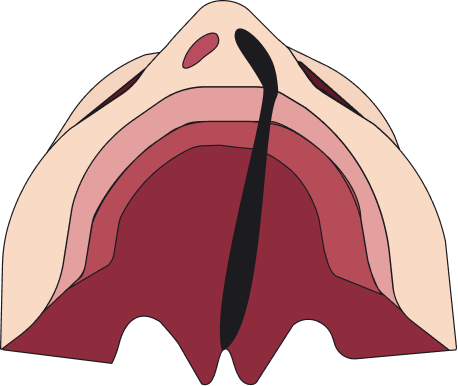
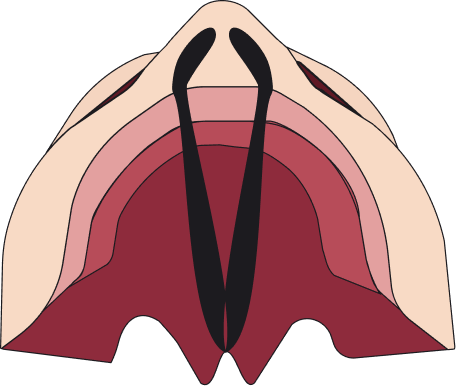
Las fisuras se pueden clasificar en 4 tipos: fisura labial unilateral, fisura labial bilateral, fisura palatina y fisura labiopalatina.

Cuando estamos en el vientre materno, todos, en algún momento, somos niños con fisura. Durante el embarazo, el rostro del bebé se forma a partir de tres partes que deben unirse entre la cuarta y la octava semana de gestación.
Cuando ese proceso no se completa, queda una pequeña separación en el labio, en el paladar o ambos. Eso es lo que se conoce como fisura.
Un niño con fisura puede tener el labio o el paladar abiertos, de manera parcial o total. Es importante saber que no falta ninguna parte de su rostro. Todas las estructuras están ahí, por lo que no es necesario un injerto, solo necesitan ser unidas mediante cirugía reconstructiva en el momento adecuado.
El primer desafío que suele presentarse al nacer un bebé con fisura es la alimentación. Puede requerir algo más de paciencia y práctica, pero con el apoyo adecuado (ya sea mediante técnicas de lactancia específicas o el uso de mamaderas adaptadas) los padres pueden alimentar a su hijo sin dificultades y con total seguridad.
Las causas de una fisura no se conoce con certeza. Se sabe que pueden influir factores hereditarios, ambientales o una mezcla de ambos, pero en la mayoría de los casos no es posible determinar una causa exacta. Lo importante es comprender que nadie tiene la culpa y que, con un tratamiento integral, los niños con fisura pueden crecer, desarrollarse y sonreír plenamente.
Cuando los padres llegan, su principal duda es cómo se va a alimentar su hijo. Piensan que se puede ahogar o que no va a poder comer.
Yo siempre les explico que el bebé no sabe que nació con fisura; quienes nos angustiamos somos nosotros. En la Fundación los ayudamos a bajar esa ansiedad, entregándoles técnicas y tipos de mamaderas que pueden usar.
Jacqueline García
Enfermera jefe de Fundación Gantz.
Un síndrome es un conjunto de signos malformativos que configuran una patología y generalmente se les denomina con el nombre del investigador de la enfermedad.
En la literatura existen alrededor de 300 síndromes que registran una fisura entre sus signos malformativos.
Desde 1985, con más de 7.200 fichas clínicas de pacientes con malformaciones craneofaciales, se ha observado que el 9% de los casos corresponde a fisuras asociadas a otra malformación o que son parte de un síndrome.
En Fundación Gantz, un 10% de las consultas corresponde a otras malformaciones craneofaciales sin fisura.
El síndrome de Adams-Oliver es un trastorno genético poco común que afecta principalmente la piel del cuero cabelludo y las extremidades.
Es muy importante comprender que la fisura labio-palatina no es una característica típica del síndrome de Adams-Oliver, aunque en ocasiones sí se han descrito malformaciones orofaciales en algunos pacientes.
Desde el nacimiento, pueden observarse zonas en el vértice del cráneo donde la piel (y a veces el hueso) no se desarrollaron correctamente. Al mismo tiempo, puede haber malformaciones en manos y pies, que van desde dedos muy cortos hasta la ausencia total de alguno de estos.
Aunque el foco central está en la piel del cuero cabelludo y las extremidades, también pueden aparecer otras complicaciones: malformaciones cardíacas, alteraciones vasculares, e incluso en algunos casos, déficits en el desarrollo neurológico.
La causa radica en alteraciones genéticas que afectan vías esenciales en la formación de vasos sanguíneos y de tejidos durante el desarrollo embrionario. Hasta ahora se han vinculado al menos seis genes.
El diagnóstico se basa en la combinación de los hallazgos de piel y extremidades, y puede confirmarse con pruebas genéticas si se encuentra la variante implicada en el síndrome.
El tratamiento es multidisciplinario, adaptado a cada persona según sus necesidades: puede involucrar cuidados de piel, cirugía reconstructiva, terapia física, evaluación cardíaca y vascular, y seguimiento neuro-lógico del paciente.
Aunque la severidad varía mucho entre los afectados, con un manejo temprano y adecuado muchas personas logran una vida plena. El pronóstico depende del grado de afectación en cada caso.
Pronóstico: generalmente bueno. La mayoría de las personas afectadas tiene una expectativa de vida normal y puede desarrollarse con autonomía, siempre que las complicaciones (en especial las cardíacas, vasculares o neurológicas) se identifiquen y traten de forma oportuna.
El Síndrome de Apert es una condición genética poco frecuente caracterizada por la fusión prematura de los huesos del cráneo (conocida como craneosinostosis) y la sindactilia o unión de los dedos de manos y pies. Esto puede generar una forma anormal de la cabeza, rasgos faciales distintivos y manos o pies con dedos fusionados, a veces en forma de “mitón”.
Está causado por mutaciones en el gen FGFR2, que regula el desarrollo de huesos y tejidos durante la gestación. Se transmite de forma autosómica dominante, aunque la mayoría de los casos ocurre de manera espontánea.
La gravedad varía de moderada a severa. El tratamiento suele implicar múltiples cirugías a lo largo de la vida para corregir la forma del cráneo, mejorar la respiración, la visión y la funcionalidad de las manos y pies. En algunos casos se observan dificultades cognitivas de grado variable, aunque también existen pacientes con inteligencia normal o superior.
En relación con la fisura labio palatina, esta puede presentarse ocasionalmente en personas con Síndrome de Apert, aunque no es un rasgo constante. Cuando aparece, se asocia principalmente a la malformación del paladar y al desarrollo anómalo de la estructura facial media, ambos casos particularmente vinculados a la craneosinostosis.
Pronóstico: depende de la severidad de las malformaciones asociadas, especialmente las que afectan al sistema nervioso central. Con un manejo quirúrgico y terapéutico adecuado, muchas personas pueden alcanzar un desarrollo favorable y una buena calidad de vida.
El síndrome EEC (Ectrodactilia-Displasia Ectodérmica-Hendidura) es una condición genética rara que puede afectar múltiples partes del cuerpo y se caracteriza por tres rasgos principales: malformaciones de las extre-midades (especialmente manos y pies), anomalías en los tejidos ectodérmicos (como piel, cabello, uñas, dientes) y fisuras orofaciales (labio y/o paladar).
Las manifestaciones clínicas pueden variar amplia-mente de una persona a otra. En cuanto a las extremidades aparece lo que se llama ectrodactilia o “split hand/split foot” (ausencia o mal desarrollo de dedos centrales, a veces denominadas “mano o pie en tenaza”). Respecto a los tejidos ectodérmicos, es frecuente encontrar cabello escaso o seco, uñas y dientes dañados o con formación alterada, sistemas de glándulas (por ejemplo sudoríparas o lagrimales) afectados, lo que puede dar sequedad de la piel, problemas de dentición o de ojos.
En relación al labio o paladar, la fisura orofacial (labio y/o paladar) está presente en un porcentaje considerable de casos (alrededor del 68% o más).
Genéticamente, la mayoría de los casos se asocian a mutaciones en el gen TP63, que codifica un factor de transcripción importante en el desarrollo de tejidos ectodérmicos y de extremidades. Este síndrome se hereda en forma autosómica dominante, aunque también pueden presentarse casos esporádicos.
El espectro de complicaciones adicionales es amplio: pueden presentarse anomalías del aparato lagrimal (estenosis u obstrucción de los conductos lagrimales), pérdida auditiva, malformaciones del tracto urinario o de los riñones, retraso de crecimiento, e incluso en un pequeño porcentaje retraso cognitivo o microcefalia.
El tratamiento es multidisciplinario: cirugía reconstructiva de la fisura orofacial, corrección de las malformaciones de manos/pies, atención dental, dermatológica, oftalmológica y apoyo funcional para la vida diaria. El manejo temprano mejora la calidad de vida.
Pronóstico: normal para el crecimiento, desarrollo y expectativas de vida en muchos casos; sin embargo, la destreza manual puede estar limitada según la extensión de la ectrodactilia, y el pronóstico funcional depende de la severidad de las manifestaciones.
El síndrome de Goldenhar es un trastorno congénito poco frecuente que afecta principalmente el desarrollo de los ojos, oídos, mandíbula y columna cervical. Se manifiesta con asimetría facial, malformaciones del oído externo y medio (que pueden causar pérdida auditiva), hipoplasia mandibular, macrostomía y dermoides o lipodermoides epibulbares (pequeños crecimientos en la superficie ocular).
La gravedad varía entre individuos: algunos presentan alteraciones leves y unilaterales, mientras que otros pueden tener compromiso en ambos lados del rostro o malformaciones adicionales en corazón, riñones o columna vertebral. Aunque no es una característica constante, puede presentarse fisura labiopalatina o paladar hendido como parte de las anomalías orofaciales vinculadas al desarrollo del primer y segundo arco branquial.
El coeficiente intelectual suele ser normal, aunque entre un 5% y 15% de los casos pueden presentar retraso del desarrollo o microcefalia, especialmente cuando hay defectos craneanos asociados. El tratamiento requiere un manejo multidisciplinario (cirugías reconstructivas, corrección auditiva, ortodoncia y apoyo terapéutico), lo que mejora notablemente la funcionalidad y la calidad de vida.
Pronóstico: generalmente bueno. En ausencia de anomalías cromosómicas o malformaciones severas, las expectativas de vida son normales.
El síndrome de Klippel-Feil es una condición congénita poco común en la que dos o más vértebras del cuello se desarrollan fusionadas desde el nacimiento.
Esta unión limita el movimiento del cuello y puede dar lugar a tres signos típicos: cuello corto, línea del cabello más baja de lo normal y dificultad para girar o mover la cabeza.
La causa se debe a una alteración en la formación de la columna cervical durante las primeras semanas del desarrollo embrionario. En la mayoría de los casos aparece de manera aislada, pero en algunos se debe a cambios genéticos (en genes como GDF6 o GDF3) que afectan la forma en que se forman los huesos y las articulaciones del cuello.
Los síntomas pueden ir de muy leves a severos. Mientras que algunas personas tienen sólo rigidez cervical, otras pueden presentar complicaciones neurológicas si la fusión vertebral provoca presión o inestabilidad en la médula espinal. Esto puede generar dolor, pérdida de fuerza, sensación de hormigueo o, en casos graves, parálisis parcial o total después de golpes o movimientos bruscos. También pueden existir problemas de audición (en alrededor del 30% de los casos), curvatura de la columna (escoliosis), o malformaciones en riñones y costillas.
Aunque el síndrome no se relaciona directamente con la fisura labiopalatina, puede coexistir con otras malformaciones faciales o del cráneo, ya que todas estas estructuras se forman a partir de los mismos tejidos embrionarios durante las primeras etapas del desarrollo del embrión.
El tratamiento requiere la participación de distintos especialistas (entre ellos traumatólogos, neurólogos, fisiatras y fonoaudiólogos) para controlar el dolor, proteger la médula espinal y mantener la movilidad. En los casos más severos se puede necesitar cirugía de estabilización del cuello.
Pronóstico: por lo general, es favorable. Las personas con formas leves o sin malformaciones adicionales pueden llevar una vida normal. Si hay complicaciones renales o neurológicas, el pronóstico depende de la atención y seguimiento médico temprano.
El síndrome de Pierre Robin es uno de los síndromes más frecuentemente asociados a la fisura palatina.
Se origina entre la quinta y sexta semana de gestación, cuando el desarrollo de la mandíbula se detiene parcial o totalmente, provocando una micrognatia (mandíbula pequeña). Al haber menos espacio en la boca, la lengua se desplaza hacia arriba, lo que impide el cierre completo del paladar entre la octava y novena semana del embarazo. Esto da lugar a una fisura palatina en forma de U.
La mandíbula pequeña también puede hacer que la lengua caiga hacia atrás (lo que se conoce como glosoptosis) y se obstruya parcialmente la faringe (la garganta), dificultando la respiración.
La severidad del cuadro puede variar. En los casos más leves, los problemas son manejables; sin embargo, en los más graves pueden presentarse dificultades para alimentarse y problemas respiratorios progresivos, llegando incluso a episodios de apnea obstructiva, que representan un riesgo vital. Por esto, el recién nacido debe ser evaluado tempranamente por un equipo especializado para establecer un diagnóstico y tratamiento adecuados.
La mayoría de los bebés con este síndrome puede ser tratada durante la etapa de recién nacido o lactante mediante medidas conservadoras, como ajustes en la posición al dormir y técnicas específicas de alimentación. Posteriormente, se realiza la cirugía para cerrar la fisura palatina, sin necesidad de intervenciones adicionales.
En casos severos de apnea obstructiva, puede requerirse una cirugía para alargar la mandíbula (distracción ósea) o, cuando no se dispone de un equipo especializado, una traqueostomía para asegurar una correcta oxigenación. Superada esta etapa, el niño tiene muy buenas posibilidades de desarrollarse normalmente.
En algunos casos puede presentarse retraso mental o daño en el sistema nervioso central, generalmente como consecuencia de la falta de oxígeno (anoxia) y las obstrucciones respiratorias durante el período neonatal.
Pronóstico: bueno, siempre que los problemas de alimentación y respiración se traten de forma temprana y efectiva.
El síndrome de Stickler es un trastorno genético del tejido conectivo que afecta principalmente los ojos, oídos, articulaciones y rasgos faciales. Esta condición se origina por mutaciones en genes que codifican colágeno (como los genes COL2A1, COL11A1 o COL11A2), lo cual altera la estructura y función del tejido de soporte en distintas partes del cuerpo.
Entre los síntomas más habituales se encuentran miopía severa (vista corta), alteraciones del humor vítreo del ojo y riesgo de desprendimiento de retina, pérdida auditiva (conductiva o sensorioneural) y problemas articulares como articulaciones muy flexibles en la infancia que luego pueden evolucionar a rigidez temprana o artrosis. También pueden presentarse rasgos faciales como mandíbula pequeña (micrognatia), paladar hendido o secuencia de Síndrome de Pierre Robin (paladar hendido, mandíbula pequeña y lengua hacia atrás) y cara aplanada.
La gravedad es muy variable: algunas personas tienen solo miopía leve y pocas molestias, mientras que otras pueden requerir múltiples intervenciones para ojos, oídos, articulaciones y alimentación/respiración en infancia. Sin embargo, el cociente intelectual no se ve directamente afectado por el síndrome.
El tratamiento es multidisciplinario: seguimiento regular por oftalmología (por el posible riesgo de desprendimiento de retina), audiología, ortopedia, cirugía maxilofacial si existe paladar hendido o mandíbula pequeña, fisioterapia/artrosis, y apoyo especializado en el desarrollo del lenguaje y la audición según sea necesario.
Pronóstico: con un manejo médico y quirúrgico adecuado, la mayoría de las personas alcanza expectativas de vida normales y buena calidad de vida. Su pronóstico funcional dependerá del grado de afectación ocular, auditiva y articular.
El síndrome de Treacher Collins es un trastorno genético que afecta principalmente el desarrollo de los huesos y tejidos de la cara.
Se caracteriza por anomalías craneofaciales como una mandíbula poco desarrollada (micrognatia), pómulos bajos o poco prominentes (hipoplasia malar), orejas pequeñas o mal formadas (microtia) y párpados que presentan una inclinación hacia abajo o incluso hendiduras en el párpado inferior (coloboma). También pueden observarse paladar hendido o fisura palatina en algunos casos.
La severidad varía mucho entre personas: en casos leves los rasgos pueden pasar casi desapercibidos, mientras que en los más complejos se requieren múltiples cirugías para corregir la estructura facial, la alimentación, la audición y la respiración.
Muchas veces se presentan también pérdida auditiva por conducto externo o medio alterados, y complicaciones respiratorias por la forma de la mandíbula o los pómulos.
El cociente intelectual suele conservarse dentro del rango normal. En algunos pocos casos (alrededor del 5 %) se ha observado retraso leve del desarrollo, aunque esto muchas veces está vinculado a la pérdida auditiva o a problemas de comunicación no tratados.
El tratamiento requiere un enfoque multidisciplinario: cirugía plástica / maxilofacial para corregir la fisonomía y facilitar la respiración y alimentación, otorrinolaringología para la audición, logopedia para el habla y seguimiento permanente. La detección temprana y la intervención son claves para lograr buena funcionalidad y calidad de vida.
Pronóstico: muy bueno. Con un diagnóstico e intervención temprano, la gran mayoría de las personas afectadas por el síndrome puede alcanzar una vida plena y con las expectativas de vida normales, siempre que se atiendan adecuadamente las complicaciones auditivas, respiratorias o de alimentación.
El síndrome de Van der Woude es un trastorno genético que se caracteriza principalmente por la presencia de hoyuelos o fositas en el labio inferior y fisuras orales, que pueden ser labiales, palatinas o ambas. Es una de las causas más frecuentes de fisura hereditaria, y suele transmitirse de forma autosómica dominante, aunque su expresión varía entre individuos de una misma familia.
Las personas afectadas pueden presentar, además de las fisuras y hoyuelos característicos, alteraciones dentales como dientes ausentes o malformados (hipodoncia), y en algunos casos anomalías del oído externo o medio, que pueden ocasionar pérdida auditiva conductiva. En menor frecuencia, se han descrito macrostomía (boca más grande de lo habitual), mandíbula pequeña o poco desarrollada (hipoplasia mandibular), y alteraciones oculares leves como dermoides epibulbares.
El manejo del síndrome requiere la participación de un equipo multidisciplinario, que puede incluir cirujanos plásticos y maxilofaciales, fonoaudiólogos, odonto-pediatras, ortodoncistas y otorrinolaringólogos. El tratamiento suele incluir cirugías reconstructivas para corregir las fisuras y mejorar la función estética y del habla, junto con seguimiento auditivo y dental para prevenir complicaciones secundarias.
Pronóstico: bueno. Las expectativas de vida son normales y el pronóstico estético y funcional suele ser muy favorable con intervención temprana. Con un acompañamiento médico adecuado, las personas con este síndrome pueden desarrollarse plenamente en lo social, educativo y emocional.
El síndrome velocardiofacial (VCFS) es causado por la pérdida de un pequeño fragmento del cromosoma 22. Esta alteración puede afectar principalmente el paladar, el corazón y la estructura facial. Entre sus características más frecuentes se encuentran la fisura palatina, las malformaciones cardíacas congénitas, la talla baja, la cabeza pequeña, la mandíbula retraída (retrognatia), la nariz alargada y las orejas pequeñas. También puede presentarse una insuficiencia velofaríngea, que dificulta el cierre adecuado entre la nariz y la boca al hablar o tragar, junto con problemas cardíacos.
La intensidad de los síntomas puede variar desde formas leves hasta moderadas. Con un diagnóstico temprano y tratamiento adecuado, el pronóstico suele ser favorable, y la mayoría de las personas afectadas puede llevar una vida plena.
Puede ocurrir un leve retraso en el desarrollo infantil. Casi todos los bebés diagnosticados presentan hipotonía (bajo tono muscular), lo que tiende a mantenerse durante los primeros meses o años de vida del infante. Más adelante, pueden aparecer dificultades de aprendizaje, especialmente en áreas como las matemáticas o comprensión lectora.
En cerca del 40% de los casos se observa un grado de discapacidad intelectual. Este impedimento, sin embargo, suele hacerse evidente al comenzar la etapa escolar, cuando las exigencias cognitivas aumentan.
En general, se observa un retraso leve en el desarrollo del lenguaje. Algunos adolescentes y adultos pueden presentar cuadros psicóticos, aunque las habilidades sociales suelen estar bien conservadas.
Pronóstico: expectativas de vida normales.
Las fisuras faciales son malformaciones congénitas que se producen en el segundo mes de embarazo, por una detención en el desarrollo del rostro del embrión en donde las estructuras que van a formar la cara no se unen, debido a factores que actualmente continúan en estudio para determinar su origen y causas.
En el 65% de los casos no se ha determinado una causa precisa para este tipo de malformación.
Según los estudios publicados el 25% de los niños fisurados tienen un antecedente familiar de fisura. El resto, no tiene causa demostrada y aún está en estudio. En estos casos se supone alguna predisposición genética sumada a algún factor ambiental.
Por el momento podemos decir que las causas de una fisura pueden ser: factores hereditarios, factores ambientales o una mezcla de ambos y que no es posible determinar con exactitud la causa específica que produjo la malformación del niño. Por lo tanto, no existe una única causa precisa que cause esta malformación.
Es un conjunto de signos clínicos que conforman un cuadro conocido, al que se le asigna un nombre. Los síndromes están estudiados y se conoce su pronóstico y tratamiento.
La fisura puede relacionarse a un síndrome, sin embargo esto no siempre ocurre.
Las fisuras no afectan el desarrollo intelectual. Por tanto, si un niño fisurado presenta una disminución intelectual, ello no es provocado por la presencia de la fisura (puede deberse a un síndrome asociado o a alguna otra alteración).
Las fisuras faciales son malformaciones congénitas.
Algunos niños con fisura tienen mayor riesgo de presentar dificultades en el aprendizaje escolar, por lo que es especialmente importante un diagnóstico y estimulación precoz de las funciones cognitivas implicadas, por parte de un especialista calificado.
En caso de que el niño presente dificultades de aprendizaje, el niño puede asistir a consultas de psicopedagogía.
En los casos de fisura de paladar blando (velo), aislada o asociada, es posible una disminución de la audición. Esto, debido a que los músculos del velo del paladar están relacionados con la función de la trompa de Eustaquio, tubo que ventila el oído medio y regula la presión que hay en él.
Si la trompa de Eustaquio no funciona adecuadamente, la presión del oído medio se mantiene negativa, lo cual provoca edema (inflamación) y acumulación de líquido llevando a otitis, la cual puede ser persistente hasta transformarse en crónica y ocasionar una disminución de la audición.
De esta manera, es de vital importancia el control periódico de la audición del niño.